
Instructor: Dean Gabriel
A few definitions:
Legitimate recombination--"homologous" recombination.
Illigitimate recombination--"nonhomologous" recombination (such as
T-DNA, IS elements and enzymatically induced, double-stranded endonuclease cuts that are then
religated, after short stretches of deletions or fill-in reactions called "nonhomologous end joining" or NHEJ)
Parasexuality--a mechanism for legitimate recombination without meiosis--occurs in viruses, bacteria and fungi. The term is, of course, most commonly applied to fungi (eukaryotes), since only eukaryotes have the option of meiosis. In some fungi, parasexuality is the predominant or only means of recombination. Parasexuality includes a system for diploid formation (plasmogamy, karyogamy in fungi), crossing over and/or gene conversion, and haploidization.
Most of the data on mitotic recombination comes from yeast, where a comparison with meiosis shows that it is much more rare than meiosis (2-3 orders of magnitude less). The fact of its rarity does not preclude the generation of linkage maps, however, although they are normalized differently (not as a percent of all progeny, because of their rarity):
For example, consider the following meiotic mapping data compared with the same data generated from mitotic recombination data in yeast.
su ribo
ad1 ad14
cen pro1
paba1 yellow
2.3
7.4 6.2
3.4 5.5
7.2 22.5
mitotic map distance
45.8 22.4
8.2 23.6
46.1 18
35.8
meiotic map distance
Note that the initiation sites or hot spots for meiotic recombination appear to be different than for mitotic recombination. Note also that most of the distances based on mitotic recombination appear to be closer (recombination is less frequent) than those obtained from meiotic mapping. However, also note that the linear order of the genes is the same.
At this point it is good to review some of the critical differences between mitosis and meiosis. Please review the Kleckner article, and note that in meiosis, after chromatid duplication, there occurs a strict alignment of homologues, with multiple interstitial interhomologue connections, all along the length of the chromatids. This duplication and alignment all occurs well prior to crossing over! If there are inversions in the homologues, the inversions cannot participate in crossing over and do not recombine, presumably because they are not aligned. Thus, initiation of recombination in meiosis requires having the homologues strictly aligned. This is not the case for mitotic recombination, in which the chromatids are clearly not aligned. Furthermore, transformation experiments using gapped plasmids have shown that integration occurs primarily at regions of homology, and the homology search takes place over the entire length of all chromosomes. Inversions do not suppress mitotic recombination or transformation. The homology search in meiosis is therefore considerably more efficient, since homologous DNA is already very close by.
Note particularly that in mitosis, there is no reductional division, but there is an equational division. Therefore, the 4N state is formed after DNA duplication, but all "Mom" chromatid centromeres separate with a "Dad" centromere, forming the 2N state again.
For a long time mitotic recombination was thought to result from crossing over, and as with meiosis, at the 4 chromatid stage, after DNA replication (it was really hard to set up the definitive three factor crosses to prove this because of the low rates of recombination). However, experimental data began accumulating, particularly from yeast studies, that demonstrated that mitotic recombination occurs predominantly by gene conversion, and also prior to DNA duplication (eg., it occurs primarily at the two chromatid stage) . This was shown using intragenic recombination with auxotrophic heteroalleles (alleles with mutational lesions in different parts of the same gene), and examination of prototrophic yeast colonies produced from single diploid cells. If single diploid cells are placed on minimal medium, rare mitotic recombinants can result in progeny cells that are prototrophic. One can isolate diploid prototrophs and then determine the markers on the other chromosome by inducing meiosis and then backcrossing the resulting progeny. In addition, not all of the cells resulting from an original single cell-derived colony are genetically identical. Sometimes prototrophic, diploid colonies produce diploid "sectors" resulting from the growth of other progeny cells that are not prototrophic. Each sector, representing a clonal lineage, can be similarly analyzed genetically in yeast by inducing meiosis and determining the resulting haploid progeny genotypes by test crosses.
Using heteroalleles at the adenine-3 and the adenine-6 genes, Hershall Roman found that prototrophic diploids never carried both mutant alleles , as would be predicted if mitotic crossing over occurred at the two chromatid stage or if crossing over were the principal means of mitotic recombination:
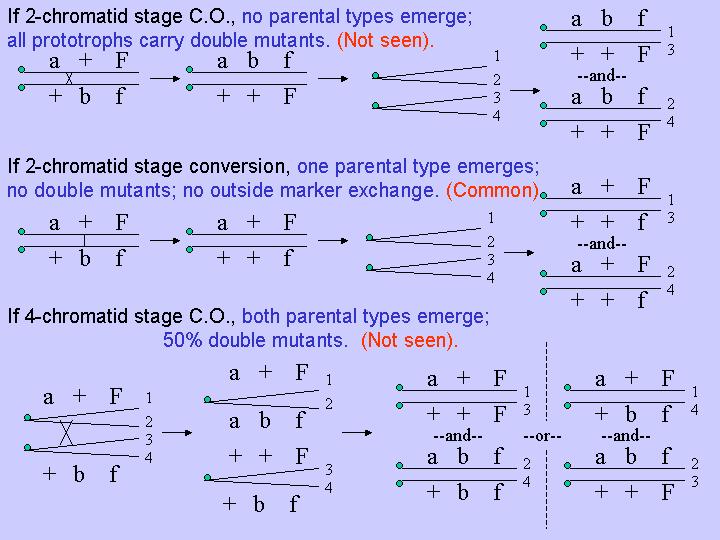
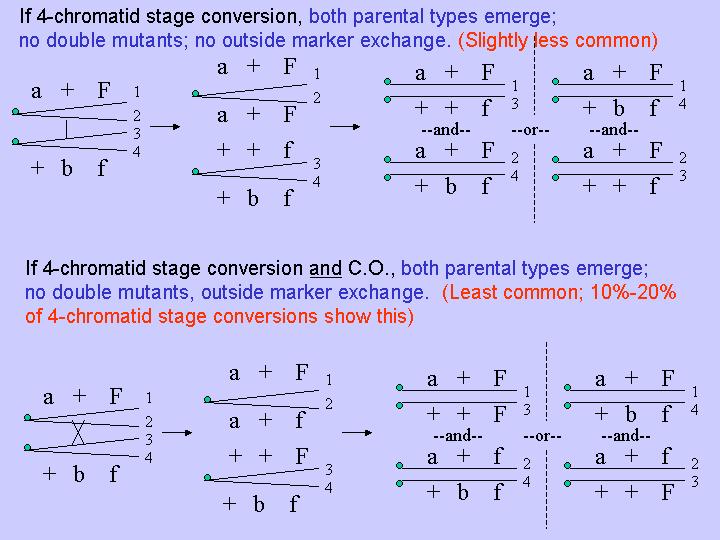
Instead, gene conversion was found to be the principal means for
mitotic recombination. In addition, mitotic gene conversion only rarely
(10% of the time) resulted in crossing over. These results indicated
that a mechanism other than double strand break repair (DSBR) was likely responsible.
Detailed analyses of the sectors resulting from single cell-derived prototrophic
colonies revealed that by far the most frequently occurring recombination
event in mitosis was gene conversion at the two chromatid stage, prior to
DNA replicatiion. The next most frequently observed result was gene
conversion at the four chromatid stage, followed by gene conversions
accompanied by crossovers (revealed by outside marker F in the above figure.
Therefore, 1) mitotic recombination occurs primarily via gene conversion;
2) gene conversion occurs primarily, but not exclusively, at the two chromatid
stage; 3) when gene conversion occurs, it can be accompanied by crossing
over, but at reduced rates compared to meiotic gene conversion..
To summarize:
Facts about Mitotic Recombination
1. Homology search covers all DNA, and alignment of chromatids does
not occur.
2. It is stimulated by double stranded breaks.
3. It is much more rare than meiotic recombination;.
4. It occurs much more frequently by gene conversion without
crossing over than by gene conversion with crossing over.
5. It occurs predominantly at the two chromatid stage, prior
to chromatid duplication and segregation to daughter cells.
How does it work?
A by now well accepted model for mitotic recombination that seems to explain the fact that mitotic recombination usually occurs without crossing over is the Synthesis-Dependent Strand Annealing (SDSA model) model. There is by now strong supporting molecular evidence for this model (for example, refer Ira et al. 2006). SDSA works similarly to the Double Strand Break Repair Model. The main difference is the strand invasion that forms the D loop results in DNA synthesis off the recipient (uncut) template (as in DSBR), BUT, there is no synthesis off of the D loop, which is sometimes called a "bubble" to indicate that it is not used as a template. A second difference is that the newly synthesized strand re-anneals to its original broken partner, without crossing over, and the D loop drops back into place (click on SDSA model for animated GIF from the Sekelsky lab (http://sekelsky.bio.unc.edu/) or refer Lecture 8 notes depicting Early Crossover Decision (ECD), NonCrossover decision pathway). In fact the SDSA model came well before the ECD model, and was adapted for use to explain noncrossovers in meiosis.
Homologous recombination is not only used by the cell in meiosis and mitosis;
as you will recall, it was found to be the
basis for integration of plasmid DNAs carrying yeast genes into the yeast
chromosome. Integration is frequently used for purposes of site-directed
mutagenesis, one of the main tools of functional genomics. Two of the
principal methods used for site directed mutagenesis are marker-interruption
and marker exchange.
To perform marker-interruption, you must clone a DNA fragment that is
entirely internal to the gene that is to be the target. If either the 5'
or 3' ends of the gene are included in the clone, the interruption won't
work, for reasons that will become apparent when you diagram the event:
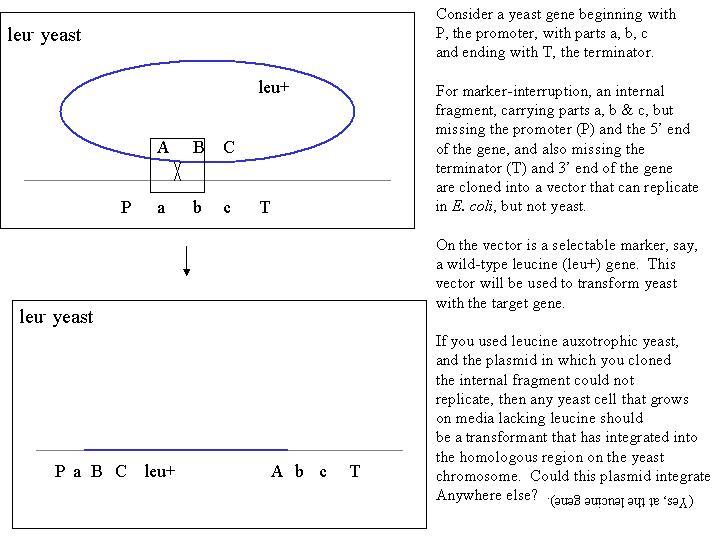
Note that the plasmid can integrate at any point that has homology, including
both the target gene, and the leucine gene.
Q: How would you ensure that the integration in yeast occurred at the
target gene only? (Hint: how was recombination
stimulated by Orr-Weaver et al? What is the mechanism of recombination???
)
Note in the above figure, there is a little black box enclosing the actual
physical exchange, which occurs by the DSBR model exactly as if the little
black box were drawn around a physical exchange region on two chromatids
in meiosis. Be sure that you can draw, using all four Watson-Crick
strands, the actual mechanism of integration, using the DSBR model. Be
sure to practice how to resolve the double Holliday structures to achieve
integration.
Prokaryotes have had to evolve a wide array of restriction endonucleases (to linearize and thus restrict double-stranded DNAs from entry),that work together with the highly efficient recBCD exonucleases (see last lecture) to protect themselves against phage infection. The recBCD exonucleases rapidly and efficiently degrade linear DNA; therefore linear DNA cannot be used to transform most prokaryotes. However, covalently closed, circular DNA can transform prokaryotes efficiently, provided the recipient strain is lacking restriction endonucleases or the covalently closed circular DNA lacks restriction endonuclease cleavage sites for those endonucleases that are produced in the recipient strain. (Phage will also produce specific methylases that protect the phage DNA from being cleaved by certain restriction enzymes.) Nevertheless, covalently closed circular DNA can even be used to transform prokaryotes by integration at reasonable efficiency, presumably because such DNA becomes (randomly) nicked, allowing binding by recA and initiating homology searching and strand invasion. Such integration requires at least 70 bp of homology in some bacteria, and much more in others, again indicating the efficiency of DNA degradation. Integration in bacteria is thought to be accomplished by double strand break repair, with the double stranded breaks resulting from paused replication forks (for more detail, refer: Lusetti, S.L. and Cox, M. M. Annual Review of Biochemistry; 2002,71:71-101),
Two methods that are widely used to make gene knockouts in bacteria depend upon reasonable efficiency of integration by covalently closed, circular plasmids that cannot replicate in the target bacteria. These methods are:
and
{kind=link}