Instructor: Dean Gabriel
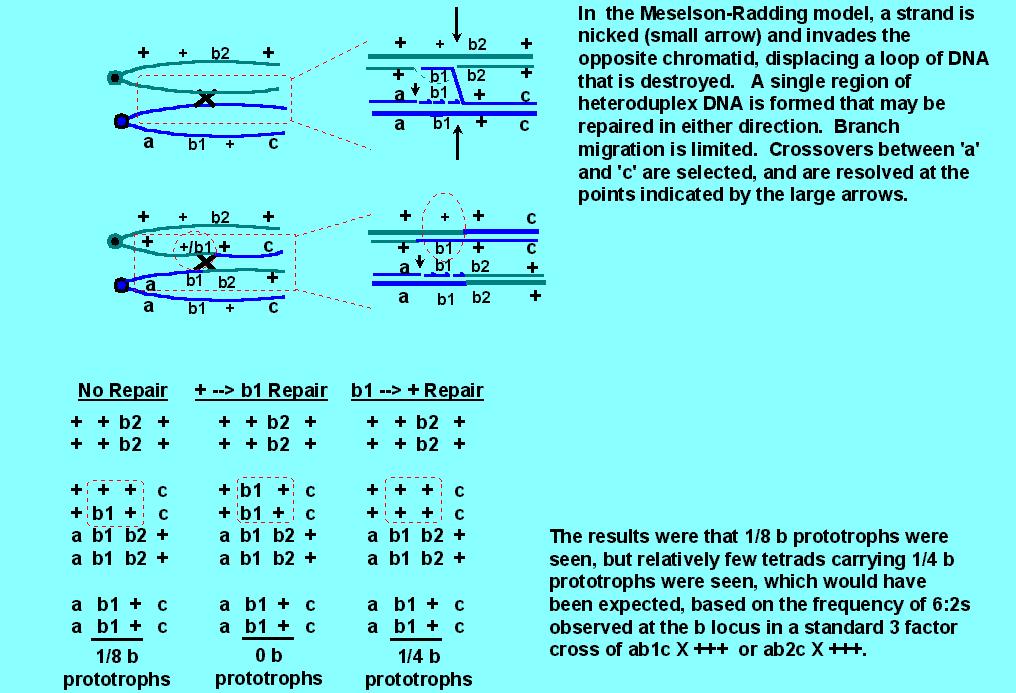
As you might imagine, the Meselson-Radding model was good, since it built on the correct Holliday foundation for resolution of crossover events, but there were some puzzling observations that it could not explain. In particular, the "default" 5:3 situation appeared to work as predicted for 5:3 tetrads (and the rare ab5:3 and ab4:4 were neatly explained by limited branch migration), but there were problems with the generation of 6:2s and 4:4s by repair that indicated that 6:2 tetrads might normally arise by an entirely different mechanism than that which gives 5:3s. The problem goes back to the observation that frequency of 6:2s > 5:3. According to the model, 6:2s arise by repair, and since 6:2s > 5:3s, repair must be efficient. If repair is efficient, one should also get approximately equal numbers of 6:2 tetrads and normal 4:4 tetrads that arise by repair. One set of observations that gave problems involved intragenic recombination in both yeast and Ascobolus. In both cases, predicted 4:4 intragenic recombination events were not explained by the Meselson-Radding model.
Consider the following cross: a b1 c X + b2 +, where b1 and b2 are two mutant alleles of a single gene, b. The mutations b1 and b2 in b are in different locations, such that recombination between the mutations can result in generation of a wild type b allele, (+). Genes 'a' and 'c' are chosen such that they are very closely linked to the b locus, so that one can look at all recombinations involving 'a' and 'c', with a reasonable expectation that the crossover event may be within 'b' (ie, intragenic recombination). It is known that 6:2s > 5:3s when the cross is a b1 c X + + + or a b2 c X + + +, indicating that corrections of b1 --> + or b2 --> + are highly efficient. But surprisingly, when the 4:4 class that results from correction of + --> b1 or + --> b2 can be observed genetically, it appears at a much lower frequency than the 5:3s. (In the diagram below, only the + --> b1 correction is illustrated, but it is representative of other data).
This indicated that either repair might not be efficient, or that the
high frequency of 6:2s could not be explained by repair; that is, that 6:2s
might be formed in a different manner from 5:3s.
At about this time, Orr-Weaver, Szostak and Rothstein (1981) published a particularly seminal paper involving transformation of yeast using plasmid DNA. Plasmids are covalently closed, circular DNAs that replicate in the bacterial cytoplasm, independently of the chromosome. In bacteria, they have "origins of replication" (for example, ori1 or colE1) which are needed to initiate replication, and antibiotic resistance genes so that their presence inside a bacterial cell (usually E. coli) can be selected on agar media containing antibiotics. Plasmids are useful for cloning (copying) genes, since some plasmids replicate to form 50-100 copies per bacterial cell, and cell density in liquid culture can reach 1010 per ml.
Eukaryotic transformations were first begun in yeast, and bacterial origins of replication are useless in eukaryotes. In fact, so are most antibiotic genes (since antibiotics are typically given to eukaryotes to fight off prokaryotic infections). If a plasmid is introduced into the cytoplasm of an eukaryotic cell, it either is degraded and lost, or it may "integrate " into the chromosome. For integration to occur without resorting to specialized pathogenic mechanisms, there must be some region of DNA sequence homology (nearly identical in DNA sequence) provided on the plasmid that is identical to what is found at some nuclear or organelle gene.
In yeast, there are many chromosomal mutations that result in auxotrophy (nutritional dependence). For example, a yeast cell with a histidine requirement cannot grow on minimal media, but requires a medium with histidine added. Some auxotrophic mutations have been well characterized and even mapped to their chromosomal locations. Since amino acid biosynthesis requires multiple enzymes in a pathway, mutations affecting one or more enzymes in the same pathway will occur in different genes. These may be numbered, as his-1, his-2 or his-3, for example, referring to different genes in the histidine biosynthetic pathway. Even alleles have been isolated, such as his-1-1, his1-2, etc.
Obviously, a copy of the wild type gene will restore prototrophy to an auxotroph. This may occur temporarily following plasmogamy (presence of the wild type copy in the same cytoplasm after cytoplasmic fusion) or permanently by karyogamy and crossing over or gene conversion. One can also provide the wild type gene by direct transformation of yeast cells. The cell wall is carefully removed by enzymes, exposing the intact membrane of the yeast "protoplast". DNA is added (usually a plasmid) that carries a gene that can serve as a selectable marker. In the case of an auxotrophic yeast cell, the wild type gene that restores prototrophy will do nicely.
Orr-Weaver et al. (1981) [PNAS 78:6354-6358] discovered that if you cut the plasmid in a yeast gene (region of homology with the recipient yeast DNA), you greatly stimulate transformation. (Cutting is achieved enzymatically, at specific known sites on the plasmid DNA, by restriction endonucleases, such as EcoRI, KpnI and BglII).
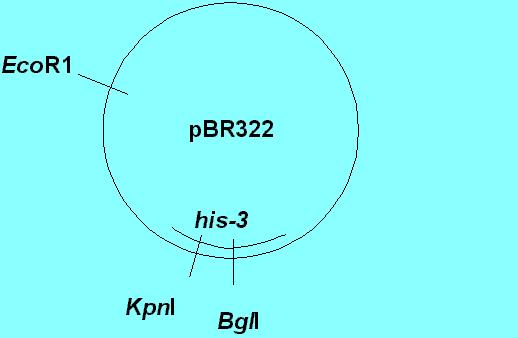
In the diagram above, which is an oversimplification of the experiments, the wild type his-3 gene from yeast was cloned into pBR322 and cloned in E. coli using ampicillin and/or tetracycline selection (both of these antibiotic resistance genes are on pBR322). The only region of homology was the his-3 gene. The essential results are as follows:
Cut site # Transformants Stimulation of transformation
Uncut
0.2 /ug DNA
----
EcoRI
3-18/ ug DNA
15-90X
KpnI
510-750/ug DNA
2,550-3750
BglII
5-450/ug DNA
25-2250
KpnI + BglII 510-750/ug
DNA 2,550-3750
Obviously, creating linear DNA by cutting the circular plasmid greatly stimulated transformation. They were also able to prove that the plasmid DNA integrated into the chromosome at a specific site that mapped to the expected his-3 gene site. Note that not only did creating a linear fragment stimulate integration at a homologous site, but that a gapped fragment also did so, and resulted in correction. This immediately stimulated model building that involved double strand breaks, and the model which is well accepted today is the Double Strand Break Repair Model.
Just as the Holliday model consistently proved reliable in predicting the resolution of crossover events, so, too, the Meselson-Radding model was proving reliable and consistent in understanding and predicting 5:3 gene conversion events. However, efficient mismatch repair, which is needed by the Meselson-Radding model to generate the 6:2s that occur in higher frequency than 5:3s, should have resulted in abundant ab4:4 tetrads that were simply not seen.
In addition, evidence was accumulating that 6:2 conversions were qualitatively, and not just quantitatively, different from 5:3 conversion events. It was clearly seen that at closely linked markers, conversions affecting one locus could also affect immediately adjacent loci (ie., co-conversion was common, just as if conversion spread from an initiation site or pole (refer below). Furthermore, not all conversion classes exhibited the same polarity; 5:3 co-conversions exhibiting polarity were not usually accompanied by 6:2 co-conversions with the same polarity, and vice versa (depending on the loci examined). Thirdly, loci exhibiting predominantly 5:3 conversions had no effect on immediately adjacent loci that showed 6:2 conversions, but those loci exhibiting predominantly 6:2 conversions had a strong effect on those showing 5:3 conversions. Taken together, this indicated that 6:2 conversions were mechanistically different from 5:3 conversions.
Let's consider some raw data involving conversions
at the grey (g) and hyaline (h) loci, which are tightly linked. h1,
h2 & h3 are heteroalleles of h (and so are all intragenic mutational
lesions).
| Class/ gene | g | h1 | h2 | h3 |
| 6m:2wt | 6 | 5 | 13 | 4 |
| 2m:6wt | 31 | 7 | 3 | 1 |
| 5m:3wt | 14 | 23 | 40 | 28 |
| 3m::5wt | 35 | 26 | 9 | 4 |
| ab4m:4wt | 13 | 35 | 35 | 16 |
| Totals: | 99 | 96 | 100 | 53 |
Second, consider the following data by G. Leblon from
Ascobolus immersus, involving two different mutations, A4 & A5
of the b2 gene. A5 is a frameshift mutation that is highly corrected,
with bias towards a correction towards the mutant genotype. A4 is a
base-substitution in the same gene, that is normally not corrected at high
frequency
| Classes/Crosses | A4+/++ | A4A5/+A5 | A4A5/++ |
| 6m:2wt | 21 | 18 | 1,900 |
| 2m:6wt | 15 | 4 | 140 |
| 5m:3wt | 103 | 108 | 371 |
| 3m::5wt | 100 | 88 | 81 |
| ab4m:4wt | 0 | 0 | 27 |
| Totals: | 2,243 | 3,189 | 28,360 |
In this table, we are looking at A4 conversions only, in the presence and absence of the A5 mutation. Note that whether or not A5 is present or absent in meiotic cells homozygous for A5, A4 has a tendency to exhibit 5:3 conversions (2nd and 3rd columns). However, when A5 is present in the heterozygous state during meiosis, there is a strong tendency for A4 to exhibit 6:2 conversions. Note also in the third column that heterozygous A5 tended to suppress the percent total of the A4 5:3s.
Is the converse true? Do loci normally showing 5:3 conversions have
an effect on loci normally showing 6:2s? The answer is plainly, no,
from the table below:
| Classes/Crosses | +A5/++ | A4A5/A4+ | A4A5/++ |
| 6m:2wt | 133 | 85 | 2,201 |
| 2m:6wt | 17 | 3 | 145 |
| 5m:3wt | 0 | 0 | 0 |
| 3m::5wt | 0 | 3 | 0 |
| ab4m:4wt | 0 | 0 | 0 |
| Totals: | 2,227 | 1,029 | 28,360 |
OK, so 6:2 conversions appeared different from 5:3 conversions, and did not seem to arise by mismatch correction. In addition, there was the curious increase in plasmid integration in yeast when linear molecules, cut in the region of homology, were used. Was there a better model? There was, called the Double-Strand Break Repair Model.
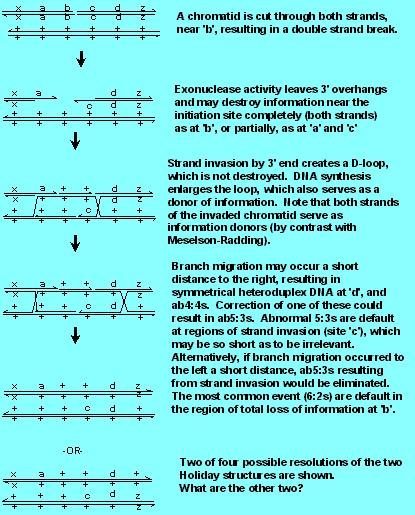
Note that the Model incorporates and preserves two aspects of the Holliday Model [(a) the generation of Holliday structures, with branch migration and symmetrical heteroduplexes, which are needed to form ab4:4s and ab5:3s and (b) resolution of gene conversions with 50% crossing over and 50% noncrossovers], one aspect of the Meselson Radding Model [strand invasion and D loop formation] but without destruction of the D loop information to give 5:3s. Note carefully how the 5:3s are generated by double strand break, since the invading strand has lost its original partner's information, while with Meselson Radding, the invading strand donates both its and its partner's information.
Finally, note that the model does not require (but there may be in some organisms) mismatch repair of any kind. 6:2s are the default of the region where there is a double strand break, 5:3s are the default of the region of strand invasion, and ab4:4s are the default of branch migration. Note that in the figure below, ab5:3s are formed at 'c' (Why are they aberrant? By definition---because there is a reciprocal exchange of information) from one chromatid to the other. Note that a normal 5:3 is formed at 'a'). There is some evidence that the "invading" 3' end can be quite short (that is, site 'c' is only rarely going to be a sizeable fraction of a gene), thus limiting the number of ab5:3s generated by this model. There are other ways to handle the problem of low numbers of ab5:3s; for example, there is nothing to prevent branch migration to the left, which would eliminate the symmetrical heteroduplex.